Test reveals patient differences in sickle cell flow
Sickle cell anemia is easy to identify but not so easy to treat. The precise conditions under which the crescent-shaped cells that characterize the disease clump together and restrict blood flow in patients have eluded scientists for as long as the disease has been documented. Therefore, whilst the last century has brought considerable advances in our molecular and genetic understanding of the disease, scientists still do not know why patients with the same genetic abnormality can present with wildly different symptoms. Recently, however, a novel diagnostic technique has shown promise in categorizing patients based on the rate at which their blood stops flowing under low oxygen levels. This tool, developed by a collaborative team of researchers from the Center for Systems Biology (MGH), Harvard, MIT and Brigham and Women’s Hospital, was reported in the March 1 issue of Science Translational Medicine.
Sickle cell anemia is caused by a single mutation in the hemoglobin molecule and is characterized by the presence of crescent (or sickle)-shaped cells with a reduced capacity for transporting oxygen around the body. Sickled red blood cells, unlike their healthy disc-shaped counterparts, are also rigid and have a propensity to clump together in capillaries, compromising blood flow. Such blockages, known as ‘crises’, cause pain and can lead to serious infections, and/or organ damage in patients.
“The big medical question,” says John Higgins, Assistant Professor of Systems Biology at MGH and a senior author on the study, “is why does a disease, that is genetically very simple, have such variable clinical consequences? We still don’t know who’s likely to have a crisis in the next month versus who might not have one for 10 years. Currently, there is no way to predict risk of crises in patients at all.” Clinical management of sickle cell anemia is thus primarily ‘after-the-fact’, targeted towards alleviating symptoms only once a crisis has occurred. In severe cases, there is an FDA-approved drug known as hydroxyurea, but it is not especially effective and has been associated with side effects.
In an effort to understand whether there is some physiological reason for this wide variability in symptom severity across patients, the collaborative research team, led by Sangeeta Bhatia at MIT, sought to simplify the blood flow anomaly that underlies sickle cell anemia. Given that low blood oxygen is one factor known to cause sickle cells to become rigid and less able to flow smoothly, the team decided to develop a microfluidic tool with which they could explore the effects of various oxygen concentrations on the flow of patient blood.
Deoxygenated sickle cells don’t go with the flow
While the initial idea for the test came from L. Mahadevan, an applied mathematician at Harvard, the microfluidic device itself was developed in Bhatia’s lab. Fabricated out of silicon, the device consisted of a system of interconnected channels: one main channel for the passage of blood and several ‘branch’ channels for gas. Minute samples of blood were injected at one end of the device and were pushed through the central channel under a constant pressure, whilst the movements of individual cells were observed and recorded via a microscope. The effects of decreasing oxygen levels on the flow of blood were subsequently assessed by injecting low oxygen gas into the branch channels, which interfaced with the main channel.
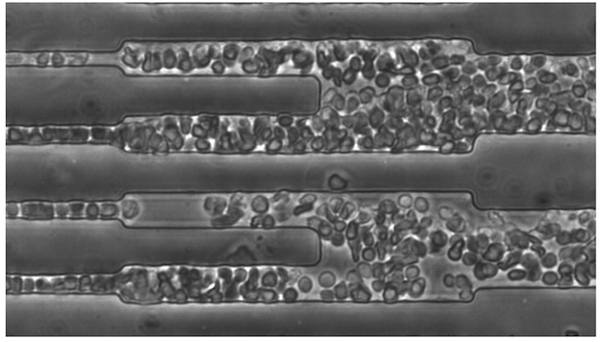
To the team’s surprise, simply deoxygenating the blood was enough to cause individual cells within the patient samples to quickly become ‘sticky’ and clumsy, clumping together as one big mass before eventually blocking the channel. Upon re-injection of oxygen, however, the cells soon reverted to their previous state of flowing easily and independently.
Based on this observation, the investigators set out to determine whether this stagnation in cell movement under low oxygen levels could be altered by increasing the pressure of blood flow. Interestingly, they found that regardless of the pressure applied to each patient sample, the time it took to stop flowing remained the same. More importantly, however, some patient’s blood appeared to stop sooner than others.
Differentiating patients
In an effort to explore the potential application of their intriguing findings, the research team performed measurements by calculating blood conductance—the rate of flow at a given pressure—under low oxygen for each sample. In so doing, they established that there were indeed patient differences. But could these variations be a indicator of disease severity?
Answering this question, however, was not straightforward since there currently exists no generally accepted categorization of sickle cell patients, against which the microfluidic test results could be assessed. “So we came up with our own categorization,” says Higgins. “We said that patients who had a crisis in the last year were ‘severe’ whereas those who hadn’t had one in at least a year were ‘benign’.” Based on this criteria, the investigators found that, in general, patients with so-defined severe disease had a much faster conductance change than patients with benign disease.
“Initially, the fact that changing the oxygen concentration stops blood flowing was just a curious physical event,” says Higgins. “What this new study is showing is that aspects of this curious physical process could be useful for categorizing patients with sickle cell disease, finding new drugs and for gaining a better understanding of the disease itself.”
Written by Yvonna Fisher-Jeffes, PhD
Wood DK, Soriano A, Mahadevan L, Higgins JM, Bhatia SN
A Biophysical Indicator of Vaso-occlusive Risk in Sickle Cell Disease.
Sci Transl Med. 2012;4(123):123ra26 – PMID: 22378926 – PMCID: PMC3633235 – Cover
{kind=link}